This function performs a standard single-cell analysis workflow.
Usage
Standard_SCP(
srt,
prefix = "Standard",
assay = NULL,
do_normalization = NULL,
normalization_method = "LogNormalize",
do_HVF_finding = TRUE,
HVF_method = "vst",
nHVF = 2000,
HVF = NULL,
do_scaling = TRUE,
vars_to_regress = NULL,
regression_model = "linear",
linear_reduction = "pca",
linear_reduction_dims = 50,
linear_reduction_dims_use = NULL,
linear_reduction_params = list(),
force_linear_reduction = FALSE,
nonlinear_reduction = "umap",
nonlinear_reduction_dims = c(2, 3),
nonlinear_reduction_params = list(),
force_nonlinear_reduction = TRUE,
neighbor_metric = "euclidean",
neighbor_k = 20L,
cluster_algorithm = "louvain",
cluster_resolution = 0.6,
seed = 11
)Arguments
- srt
A Seurat object.
- prefix
A prefix to add to the names of intermediate objects created by the function (default is "Standard").
- assay
The name of the assay to use for the analysis. If NULL, the default assay of the Seurat object will be used.
- do_normalization
A logical value indicating whether to perform normalization. If NULL, normalization will be performed if the specified assay does not have scaled data.
- normalization_method
The method to use for normalization. Options are "LogNormalize", "SCT", or "TFIDF" (default is "LogNormalize").
- do_HVF_finding
A logical value indicating whether to perform high variable feature finding. If TRUE, the function will force to find the highly variable features (HVF) using the specified HVF method.
- HVF_method
The method to use for finding highly variable features. Options are "vst", "mvp" or "disp" (default is "vst").
- nHVF
The number of highly variable features to select. If NULL, all highly variable features will be used.
- HVF
A vector of feature names to use as highly variable features. If NULL, the function will use the highly variable features identified by the HVF method.
- do_scaling
A logical value indicating whether to perform scaling. If TRUE, the function will force to scale the data using the ScaleData function.
- vars_to_regress
A vector of feature names to use as regressors in the scaling step. If NULL, no regressors will be used.
- regression_model
The regression model to use for scaling. Options are "linear", "poisson", or "negativebinomial" (default is "linear").
- linear_reduction
The linear dimensionality reduction method to use. Options are "pca", "svd", "ica", "nmf", "mds", or "glmpca" (default is "pca").
- linear_reduction_dims
The number of dimensions to keep after linear dimensionality reduction (default is 50).
- linear_reduction_dims_use
The dimensions to use for downstream analysis. If NULL, all dimensions will be used.
- linear_reduction_params
A list of parameters to pass to the linear dimensionality reduction method.
- force_linear_reduction
A logical value indicating whether to force linear dimensionality reduction even if the specified reduction is already present in the Seurat object.
- nonlinear_reduction
The nonlinear dimensionality reduction method to use. Options are "umap","umap-naive", "tsne", "dm", "phate", "pacmap", "trimap", "largevis", or "fr" (default is "umap").
- nonlinear_reduction_dims
The number of dimensions to keep after nonlinear dimensionality reduction. If a vector is provided, different numbers of dimensions can be specified for each method (default is c(2, 3)).
- nonlinear_reduction_params
A list of parameters to pass to the nonlinear dimensionality reduction method.
- force_nonlinear_reduction
A logical value indicating whether to force nonlinear dimensionality reduction even if the specified reduction is already present in the Seurat object.
- neighbor_metric
The distance metric to use for finding neighbors. Options are "euclidean", "cosine", "manhattan", or "hamming" (default is "euclidean").
- neighbor_k
The number of nearest neighbors to use for finding neighbors (default is 20).
- cluster_algorithm
The clustering algorithm to use. Options are "louvain", "slm", or "leiden" (default is "louvain").
- cluster_resolution
The resolution parameter to use for clustering. Larger values result in fewer clusters (default is 0.6).
- seed
The random seed to use for reproducibility (default is 11).
Examples
data("pancreas_sub")
pancreas_sub <- Standard_SCP(pancreas_sub)
#> [2025-09-08 16:17:40.841751] Start Standard_SCP
#> [2025-09-08 16:17:40.84197] Checking srtList... ...
#> Warning: Failed to check Seurat version compatibility: 'list' object cannot be coerced to type 'double'
#> Warning: The following arguments are not used: drop
#> Warning: The following arguments are not used: drop
#> Error in as.vector(data): no method for coercing this S4 class to a vector
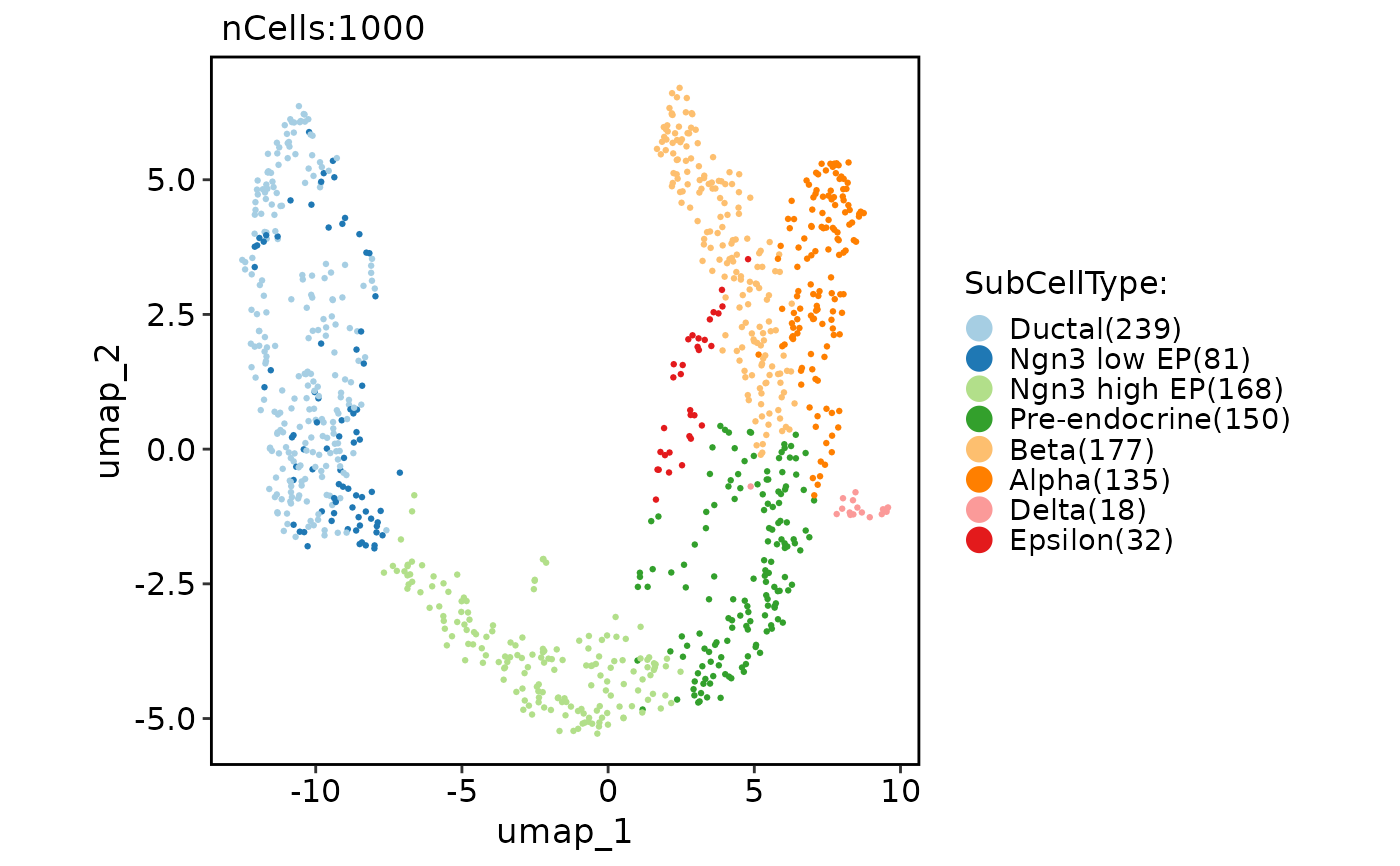
CellDimPlot(pancreas_sub, group.by = "SubCellType")
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
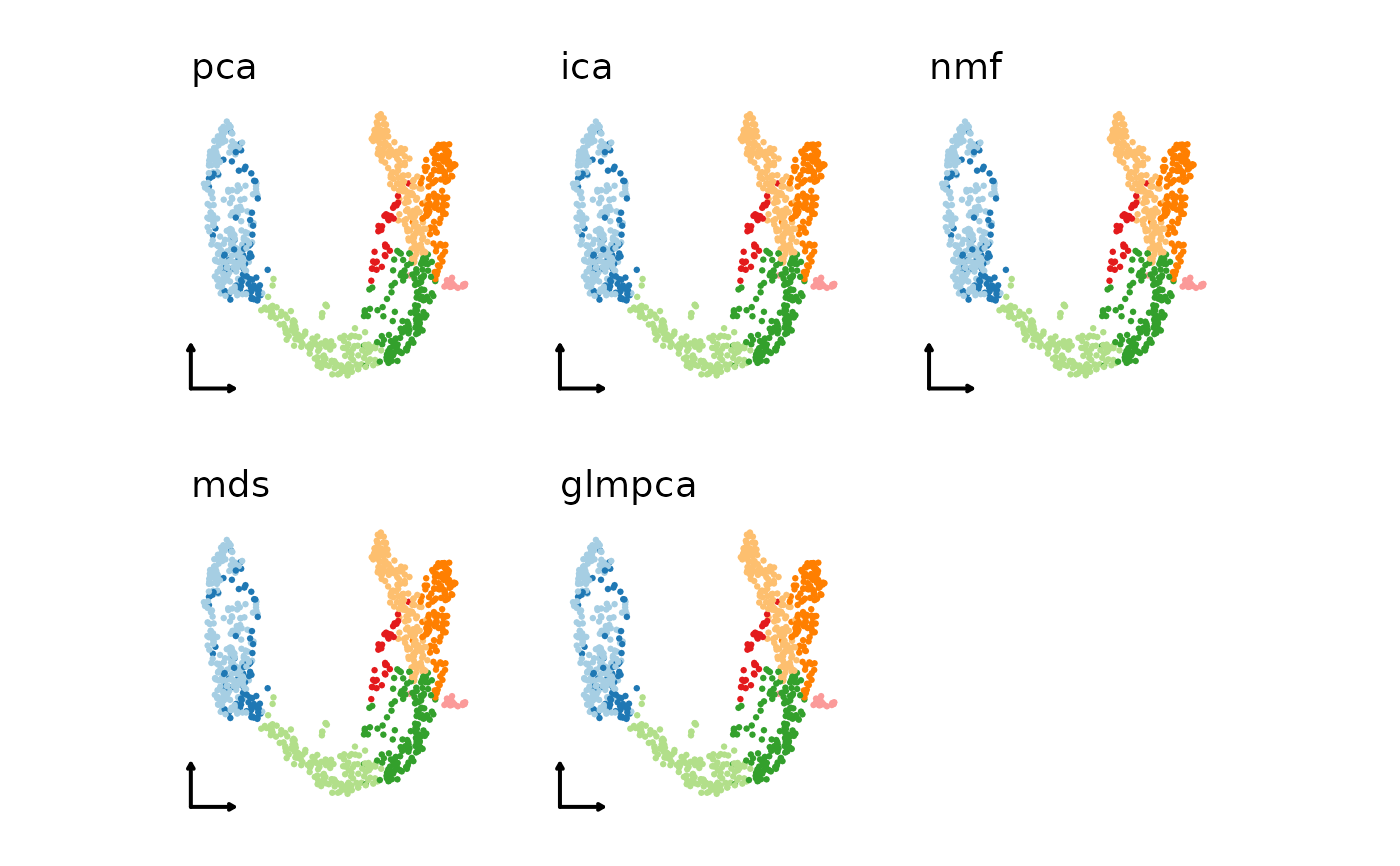
 # Use a combination of different linear or non-linear dimension reduction methods
linear_reductions <- c("pca", "ica", "nmf", "mds", "glmpca")
pancreas_sub <- Standard_SCP(
pancreas_sub,
linear_reduction = linear_reductions,
nonlinear_reduction = "umap"
)
#> [2025-09-08 16:17:41.487652] Start Standard_SCP
#> [2025-09-08 16:17:41.487813] Checking srtList... ...
#> Warning: Failed to check Seurat version compatibility: 'list' object cannot be coerced to type 'double'
#> Warning: The following arguments are not used: drop
#> Warning: The following arguments are not used: drop
#> Error in as.vector(data): no method for coercing this S4 class to a vector
plist1 <- lapply(linear_reductions, function(lr) {
CellDimPlot(pancreas_sub,
group.by = "SubCellType",
reduction = paste0("Standard", lr, "UMAP2D"),
xlab = "", ylab = "", title = lr,
legend.position = "none",
theme_use = "theme_blank"
)
})
patchwork::wrap_plots(plotlist = plist1)
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
# Use a combination of different linear or non-linear dimension reduction methods
linear_reductions <- c("pca", "ica", "nmf", "mds", "glmpca")
pancreas_sub <- Standard_SCP(
pancreas_sub,
linear_reduction = linear_reductions,
nonlinear_reduction = "umap"
)
#> [2025-09-08 16:17:41.487652] Start Standard_SCP
#> [2025-09-08 16:17:41.487813] Checking srtList... ...
#> Warning: Failed to check Seurat version compatibility: 'list' object cannot be coerced to type 'double'
#> Warning: The following arguments are not used: drop
#> Warning: The following arguments are not used: drop
#> Error in as.vector(data): no method for coercing this S4 class to a vector
plist1 <- lapply(linear_reductions, function(lr) {
CellDimPlot(pancreas_sub,
group.by = "SubCellType",
reduction = paste0("Standard", lr, "UMAP2D"),
xlab = "", ylab = "", title = lr,
legend.position = "none",
theme_use = "theme_blank"
)
})
patchwork::wrap_plots(plotlist = plist1)
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
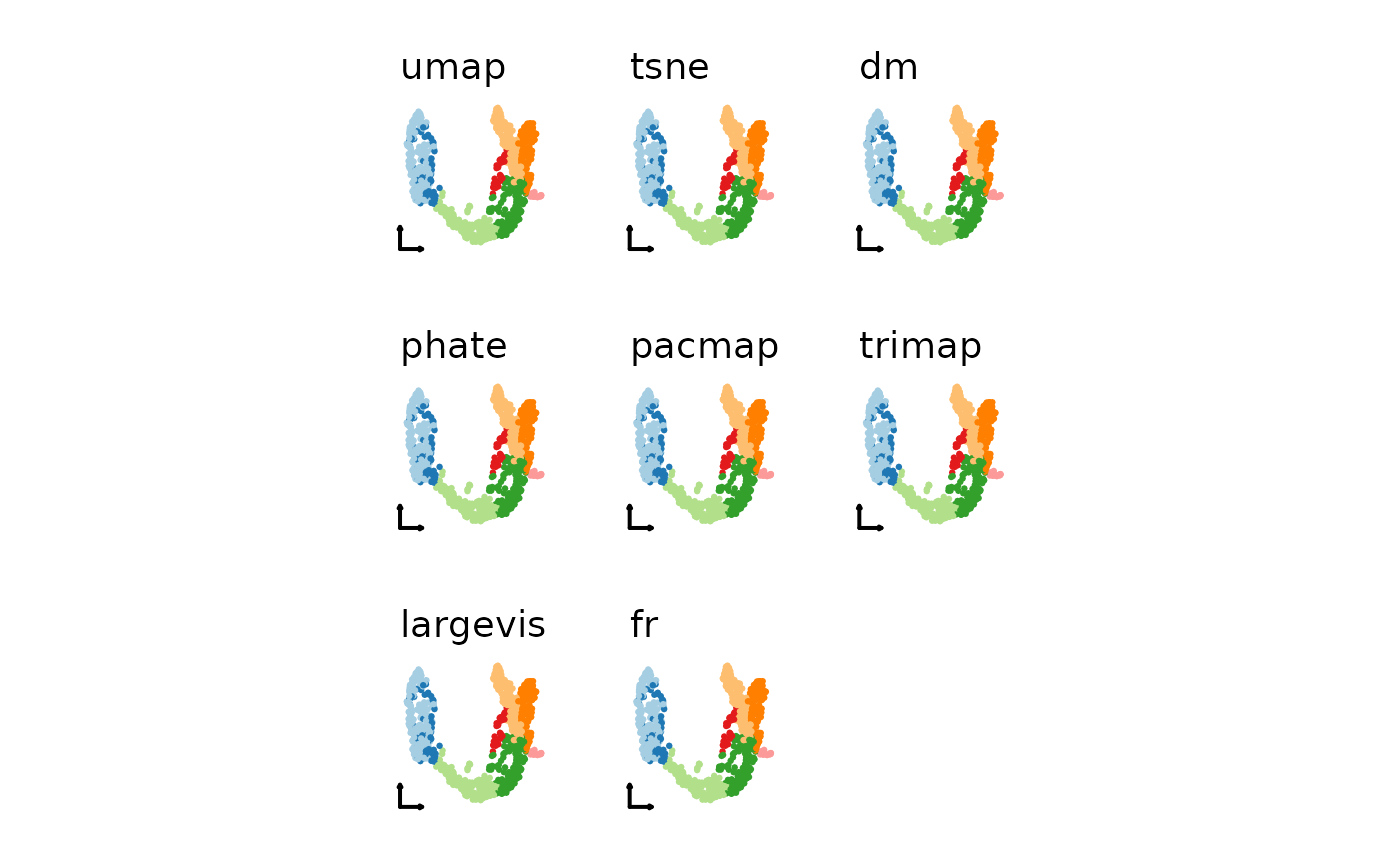
 nonlinear_reductions <- c("umap", "tsne", "dm", "phate", "pacmap", "trimap", "largevis", "fr")
pancreas_sub <- Standard_SCP(
pancreas_sub,
linear_reduction = "pca",
nonlinear_reduction = nonlinear_reductions
)
#> [2025-09-08 16:17:42.678782] Start Standard_SCP
#> [2025-09-08 16:17:42.678946] Checking srtList... ...
#> Warning: Failed to check Seurat version compatibility: 'list' object cannot be coerced to type 'double'
#> Warning: The following arguments are not used: drop
#> Warning: The following arguments are not used: drop
#> Error in as.vector(data): no method for coercing this S4 class to a vector
plist2 <- lapply(nonlinear_reductions, function(nr) {
CellDimPlot(pancreas_sub,
group.by = "SubCellType",
reduction = paste0("Standardpca", toupper(nr), "2D"),
xlab = "", ylab = "", title = nr,
legend.position = "none",
theme_use = "theme_blank"
)
})
patchwork::wrap_plots(plotlist = plist2)
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
nonlinear_reductions <- c("umap", "tsne", "dm", "phate", "pacmap", "trimap", "largevis", "fr")
pancreas_sub <- Standard_SCP(
pancreas_sub,
linear_reduction = "pca",
nonlinear_reduction = nonlinear_reductions
)
#> [2025-09-08 16:17:42.678782] Start Standard_SCP
#> [2025-09-08 16:17:42.678946] Checking srtList... ...
#> Warning: Failed to check Seurat version compatibility: 'list' object cannot be coerced to type 'double'
#> Warning: The following arguments are not used: drop
#> Warning: The following arguments are not used: drop
#> Error in as.vector(data): no method for coercing this S4 class to a vector
plist2 <- lapply(nonlinear_reductions, function(nr) {
CellDimPlot(pancreas_sub,
group.by = "SubCellType",
reduction = paste0("Standardpca", toupper(nr), "2D"),
xlab = "", ylab = "", title = nr,
legend.position = "none",
theme_use = "theme_blank"
)
})
patchwork::wrap_plots(plotlist = plist2)
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.
#> Warning: No shared levels found between `names(values)` of the manual scale and the data's fill values.