Immune Repertoire Metadata Preservation
Source:vignettes/immune-repertoire.Rmd
immune-repertoire.RmdIntroduction
Single-cell immune profiling experiments often pair gene expression with TCR/BCR sequencing. Tools like scRepertoire add clonotype annotations – clonotype IDs, V/D/J gene usage, CDR3 sequences, and clone frequencies – as cell-level metadata. scConvert preserves all of this metadata through format conversions, so TCR/BCR annotations survive roundtrips between Seurat and h5ad.
Load PBMC data and add synthetic TCR metadata
We start with the shipped PBMC demo (500 cells, 9 cell types) and add synthetic TCR annotations to demonstrate metadata preservation.
pbmc_path <- system.file("extdata", "pbmc_demo.rds", package = "scConvert")
obj <- readRDS(pbmc_path)
cat("Cells:", ncol(obj), "\n")
#> Cells: 500
cat("Genes:", nrow(obj), "\n")
#> Genes: 2000
cat("Cell types:", paste(levels(obj$seurat_annotations), collapse = ", "), "\n")
#> Cell types: Naive CD4 T, Memory CD4 T, CD14+ Mono, B, CD8 T, FCGR3A+ Mono, NK, DC, PlateletAdd TCR annotations
In a real workflow, these columns would come from scRepertoire’s
combineExpression(). Here we create synthetic clonotype
data to illustrate the conversion.
set.seed(42)
n_cells <- ncol(obj)
# 8 clonotypes with power-law-like frequencies
clonotype_ids <- paste0("clonotype_", sprintf("%03d", 1:8))
weights <- c(30, 22, 16, 12, 8, 5, 4, 3)
assignments <- sample(seq_along(clonotype_ids), n_cells,
replace = TRUE, prob = weights)
obj$clonotype_id <- factor(clonotype_ids[assignments], levels = clonotype_ids)
# V/J gene usage
tra_genes <- c("TRAV1-2", "TRAV12-1", "TRAV38-2", "TRAV21",
"TRAV8-6", "TRAV26-1", "TRAV13-1", "TRAV29")
trb_genes <- c("TRBV6-1", "TRBV28", "TRBV5-1", "TRBV7-2",
"TRBV19", "TRBV12-3", "TRBV4-1", "TRBV20-1")
obj$tra_gene <- tra_genes[assignments]
obj$trb_gene <- trb_genes[assignments]
# Clone frequency
freq_tbl <- table(assignments)
obj$clone_frequency <- as.integer(freq_tbl[as.character(assignments)])
cat("TCR columns added:", paste(c("clonotype_id", "tra_gene",
"trb_gene", "clone_frequency"), collapse = ", "), "\n")
#> TCR columns added: clonotype_id, tra_gene, trb_gene, clone_frequency
Convert to h5ad and read back
h5ad_file <- tempfile(fileext = ".h5ad")
writeH5AD(obj, h5ad_file, overwrite = TRUE)
obj_rt <- readH5AD(h5ad_file, verbose = FALSE)Verify TCR metadata preservation
tcr_cols <- c("clonotype_id", "tra_gene", "trb_gene", "clone_frequency")
cat("All TCR columns present:",
all(tcr_cols %in% colnames(obj_rt[[]])), "\n")
#> All TCR columns present: TRUE
cat("clonotype_id match:",
all(as.character(obj$clonotype_id) ==
as.character(obj_rt$clonotype_id)), "\n")
#> clonotype_id match: TRUE
cat("tra_gene match:", all(obj$tra_gene == obj_rt$tra_gene), "\n")
#> tra_gene match: TRUE
cat("trb_gene match:", all(obj$trb_gene == obj_rt$trb_gene), "\n")
#> trb_gene match: TRUE
cat("clone_frequency match:",
all(obj$clone_frequency == obj_rt$clone_frequency), "\n")
#> clone_frequency match: TRUEAll TCR metadata columns – factors, strings, and integers – are preserved exactly through the h5ad roundtrip.
Clonotype frequency barplot
freq_df <- as.data.frame(table(obj_rt$clonotype_id))
colnames(freq_df) <- c("Clonotype", "Cells")
freq_df <- freq_df[order(-freq_df$Cells), ]
barplot(freq_df$Cells, names.arg = freq_df$Clonotype,
col = "steelblue", las = 2, cex.names = 0.8,
main = "Clonotype frequencies (after roundtrip)",
ylab = "Number of cells")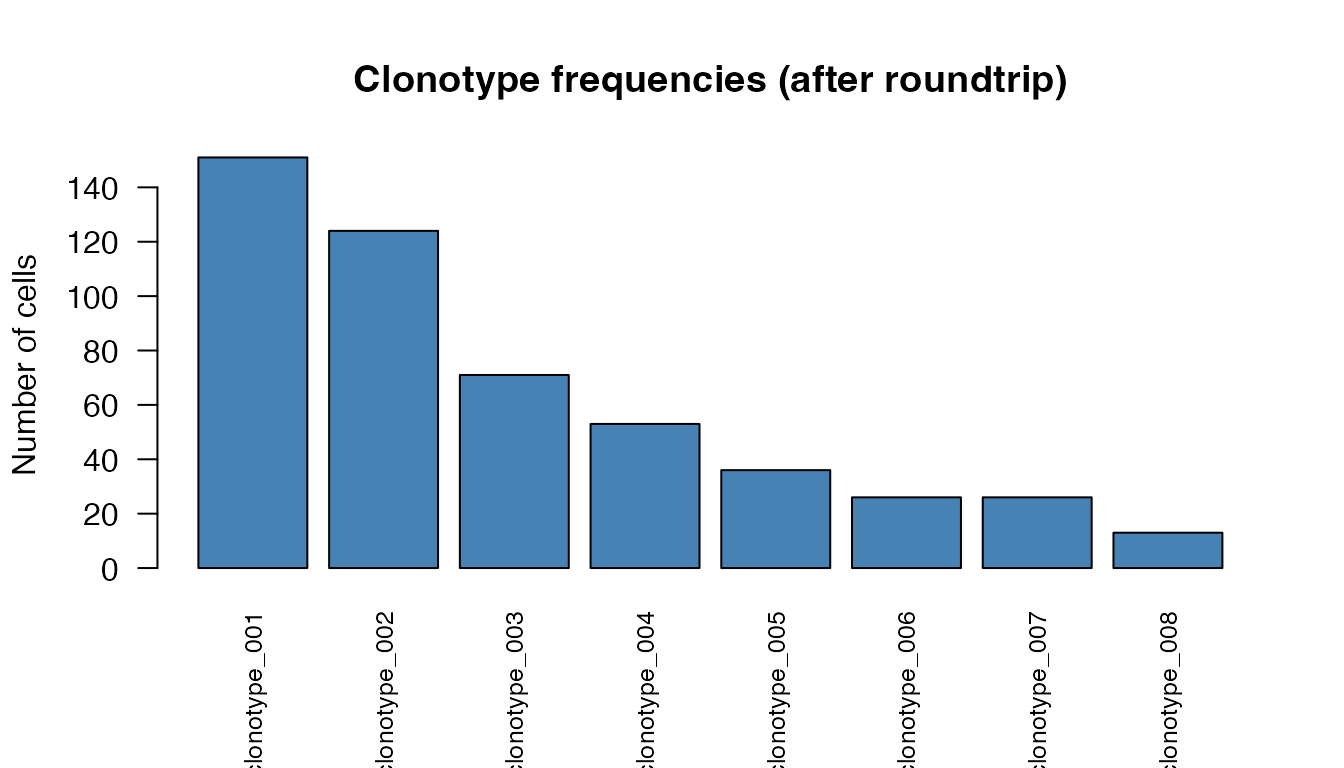
Gene expression across cell types
Cell type annotations and expression values are also preserved alongside the TCR metadata.
VlnPlot(obj_rt, features = "LYZ", group.by = "seurat_annotations",
pt.size = 0.1) +
ggplot2::ggtitle("LYZ expression by cell type (after roundtrip)") +
ggplot2::theme(legend.position = "none")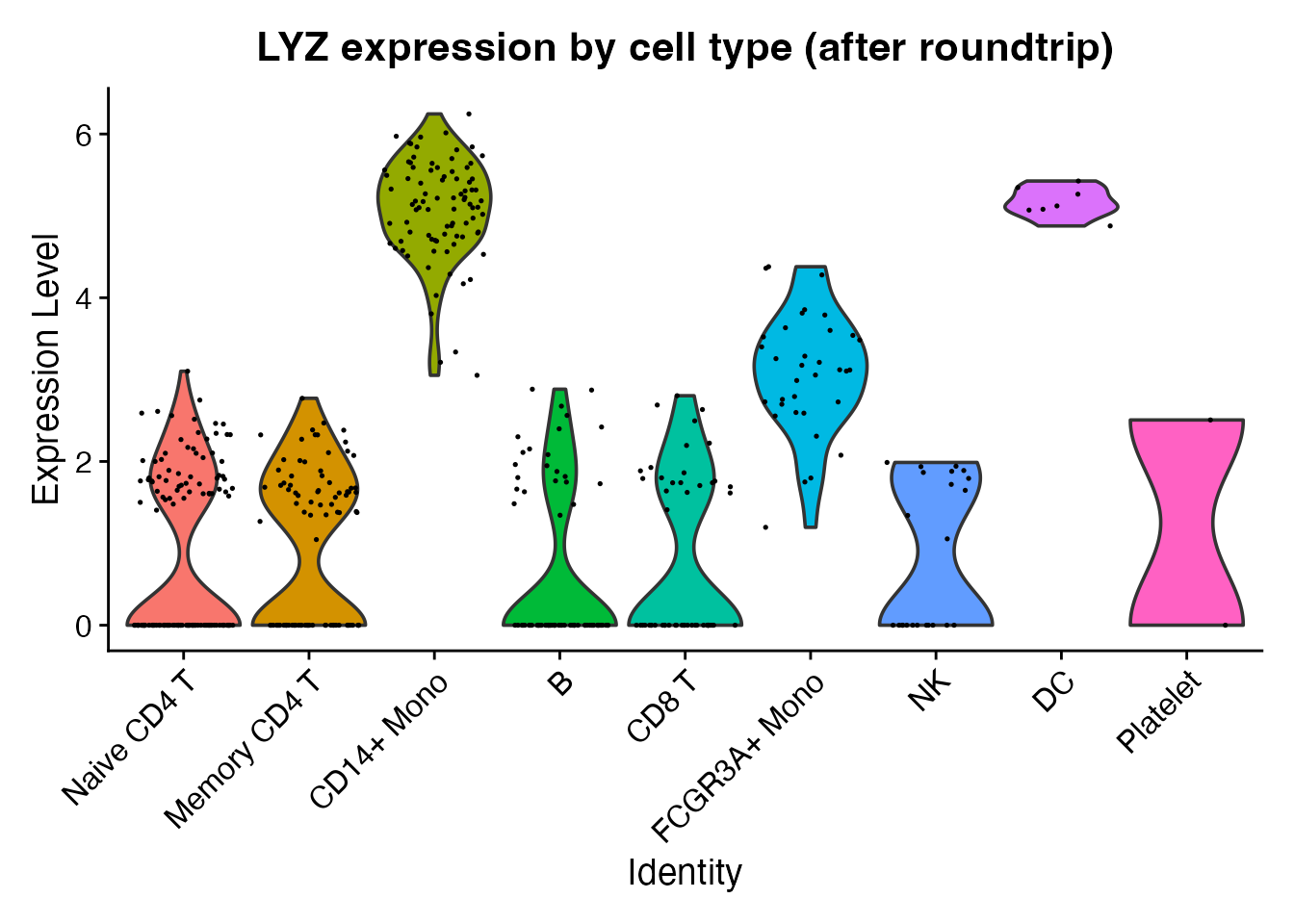
Session Info
sessionInfo()
#> R version 4.6.0 (2026-04-24)
#> Platform: x86_64-pc-linux-gnu
#> Running under: Ubuntu 24.04.4 LTS
#>
#> Matrix products: default
#> BLAS: /usr/lib/x86_64-linux-gnu/openblas-pthread/libblas.so.3
#> LAPACK: /usr/lib/x86_64-linux-gnu/openblas-pthread/libopenblasp-r0.3.26.so; LAPACK version 3.12.0
#>
#> locale:
#> [1] LC_CTYPE=C.UTF-8 LC_NUMERIC=C LC_TIME=C.UTF-8
#> [4] LC_COLLATE=C.UTF-8 LC_MONETARY=C.UTF-8 LC_MESSAGES=C.UTF-8
#> [7] LC_PAPER=C.UTF-8 LC_NAME=C LC_ADDRESS=C
#> [10] LC_TELEPHONE=C LC_MEASUREMENT=C.UTF-8 LC_IDENTIFICATION=C
#>
#> time zone: UTC
#> tzcode source: system (glibc)
#>
#> attached base packages:
#> [1] stats graphics grDevices utils datasets methods base
#>
#> other attached packages:
#> [1] ggplot2_4.0.3 Seurat_5.5.0 SeuratObject_5.4.0 sp_2.2-1
#> [5] scConvert_0.2.0
#>
#> loaded via a namespace (and not attached):
#> [1] RColorBrewer_1.1-3 jsonlite_2.0.0 magrittr_2.0.5
#> [4] spatstat.utils_3.2-2 farver_2.1.2 rmarkdown_2.31
#> [7] fs_2.1.0 ragg_1.5.2 vctrs_0.7.3
#> [10] ROCR_1.0-12 spatstat.explore_3.8-0 htmltools_0.5.9
#> [13] sass_0.4.10 sctransform_0.4.3 parallelly_1.47.0
#> [16] KernSmooth_2.23-26 bslib_0.10.0 htmlwidgets_1.6.4
#> [19] desc_1.4.3 ica_1.0-3 plyr_1.8.9
#> [22] plotly_4.12.0 zoo_1.8-15 cachem_1.1.0
#> [25] igraph_2.3.0 mime_0.13 lifecycle_1.0.5
#> [28] pkgconfig_2.0.3 Matrix_1.7-5 R6_2.6.1
#> [31] fastmap_1.2.0 MatrixGenerics_1.24.0 fitdistrplus_1.2-6
#> [34] future_1.70.0 shiny_1.13.0 digest_0.6.39
#> [37] S4Vectors_0.50.0 patchwork_1.3.2 tensor_1.5.1
#> [40] RSpectra_0.16-2 irlba_2.3.7 GenomicRanges_1.64.0
#> [43] textshaping_1.0.5 labeling_0.4.3 progressr_0.19.0
#> [46] spatstat.sparse_3.1-0 httr_1.4.8 polyclip_1.10-7
#> [49] abind_1.4-8 compiler_4.6.0 bit64_4.8.0
#> [52] withr_3.0.2 S7_0.2.2 fastDummies_1.7.6
#> [55] MASS_7.3-65 tools_4.6.0 lmtest_0.9-40
#> [58] otel_0.2.0 httpuv_1.6.17 future.apply_1.20.2
#> [61] goftest_1.2-3 glue_1.8.1 nlme_3.1-169
#> [64] promises_1.5.0 grid_4.6.0 Rtsne_0.17
#> [67] cluster_2.1.8.2 reshape2_1.4.5 generics_0.1.4
#> [70] hdf5r_1.3.12 gtable_0.3.6 spatstat.data_3.1-9
#> [73] tidyr_1.3.2 data.table_1.18.2.1 BiocGenerics_0.58.0
#> [76] BPCells_0.3.1 spatstat.geom_3.7-3 RcppAnnoy_0.0.23
#> [79] ggrepel_0.9.8 RANN_2.6.2 pillar_1.11.1
#> [82] stringr_1.6.0 spam_2.11-3 RcppHNSW_0.6.0
#> [85] later_1.4.8 splines_4.6.0 dplyr_1.2.1
#> [88] lattice_0.22-9 survival_3.8-6 bit_4.6.0
#> [91] deldir_2.0-4 tidyselect_1.2.1 miniUI_0.1.2
#> [94] pbapply_1.7-4 knitr_1.51 gridExtra_2.3
#> [97] Seqinfo_1.2.0 IRanges_2.46.0 scattermore_1.2
#> [100] stats4_4.6.0 xfun_0.57 matrixStats_1.5.0
#> [103] stringi_1.8.7 lazyeval_0.2.3 yaml_2.3.12
#> [106] evaluate_1.0.5 codetools_0.2-20 tibble_3.3.1
#> [109] cli_3.6.6 uwot_0.2.4 xtable_1.8-8
#> [112] reticulate_1.46.0 systemfonts_1.3.2 jquerylib_0.1.4
#> [115] Rcpp_1.1.1-1.1 globals_0.19.1 spatstat.random_3.4-5
#> [118] png_0.1-9 spatstat.univar_3.1-7 parallel_4.6.0
#> [121] pkgdown_2.2.0 dotCall64_1.2 listenv_0.10.1
#> [124] viridisLite_0.4.3 scales_1.4.0 ggridges_0.5.7
#> [127] purrr_1.2.2 crayon_1.5.3 rlang_1.2.0
#> [130] cowplot_1.2.0